- Nanocristal (à gauche) et poudre (à droite).
- Les mesures expérimentales indiquent que les nanocristaux sont créés dans
la phase gazeuse à partir d'une certaine pression car des substrats de différentes
températures donnent des spectres
Raman différents. Cette différence de résultats
peut être comprise en prenant en compte la force de thermophorèse dans le
processus de dépôt.
- L'accroissement de la puissance de calcul suit une loi empirique de
doublement de la puissance tous les un an et demi. Nous représentons ici le
nombre d'opérations à virgule flottante par seconde (flops)
des supercalculateurs en fonction de l'année de leur création.
- L'erreur commise sur l'énergie dépend de la précision machine.
En effet, une modélisation basée sur l'utilisation de nombres qui n'ont pas une bonne précision
donne une trajectoire incorrecte lorsque le pas de temps est trop petit.
- Visualisation graphique de l'approximation effectuée dans l'algorithme d'Euler.
- Visualisation graphique de l'approximation effectuée dans l'algorithme de Runge-Kutta.
- L'erreur pour un algorithme de Gear est bien moins importante que celle
d'un algorithme de Verlet. Le pas de temps ainsi que la précision sont en unités réduites.
- Exemple de calcul d'orbitale moléculaire dans le cas d'une molécule de silane.
Les orbitales sont représentées selon leur nombre quantique principale 'n' et leur nombre
quantique angulaire 'l'. Nous donnons aussi la valeur énergétique de l'orbitale en unité atomique.
- Niveaux d'énergie de la molécule AB. Nous remarquons la formation d'une orbitale
liante et d'une orbitale anti-liante. L'orbitale liante a un niveau plus bas en énergie
que l'orbitale anti-liante. Les deux électrons de A se placent donc sur le niveau de plus
basse énergie pour former la liaison AB.
- Chaîne parfaitement alternée obtenue lorsque
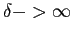 .
.
- Valeurs propres à chaque nouvel atome collé. Nous noterons que pour AB,
c'est-à-dire pour une chaîne de taille 2 nous formons les deux niveaux liant
et anti-liant.
Le niveau de Fermi (en rouge)
monte lorsque nous collons un atome A (qui possède deux électrons) et descend
lorsque nous collons un atome B. Nous pouvons donc voir que cette chaîne s'est auto-organisée en
ABABAABABABABBABABAB...
- Les retournements de blocs sont de moins en moins fréquents lorsque nous nous
éloignons de la perturbation initiale (germe de croissance AAAAAAAAAA)
- Comparaison de courbes de potentiel pour la liaison
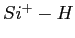 , obtenues suivant
différentes méthodes.
, obtenues suivant
différentes méthodes.
- Surface de potentiel pour la molécule
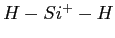 calculé avec la
méthode PM3.
calculé avec la
méthode PM3.
- Surface de potentiel pour la molécule
calculé avec la
méthode "coupled clusters".
- Configuration géométrique de minimum d'énergie potentielle obtenue en simulation
avec des potentiels calculés par une méthode semi-empirique de type PM3.
- Section efficace de production des molécules issues de la réaction
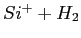 .
La ligne continue correspond aux résultats expérimentaux et les étoiles (cercles) aux valeurs
obtenues en simulation de la dynamique moléculaire de la réaction.
.
La ligne continue correspond aux résultats expérimentaux et les étoiles (cercles) aux valeurs
obtenues en simulation de la dynamique moléculaire de la réaction.
- Vérification de la section efficace de production du
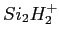 issue de la réaction
issue de la réaction
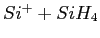 .
La ligne continue correspond aux résultats expérimentaux, et les étoiles aux valeurs
obtenues par la simulation de la dynamique moléculaire de la réaction.
Les statistiques sont obtenues après la simulation de près de 10000 trajectoires.
.
La ligne continue correspond aux résultats expérimentaux, et les étoiles aux valeurs
obtenues par la simulation de la dynamique moléculaire de la réaction.
Les statistiques sont obtenues après la simulation de près de 10000 trajectoires.
- Profil de potentiel moyenné dans le temps.
- Profil d'énergie des ions moyenné dans le temps.
- Profil d'énergie des ions moyenné dans le temps.
- Distribution de probabilité des vitesses des molécules dans le plasma.
- Illustration d'un exemple typique de mécanisme de croissance. Nous initialisons la croissance
avec un radical
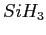 . Des réactions successives avec du
. Des réactions successives avec du 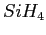 permettent la croissance
d'un agrégat.
permettent la croissance
d'un agrégat.
- Exemple d'agrégat amorphe obtenu par une simulation de croissance à température
ambiante dans un plasma de silane pur.
- Nous constatons un très bon accord entre les structures de minimum d'énergie obtenues par calcul
ab-initio et les structures formées lors de croissances à haute température avec la méthode PM3.
- Le battement observé est une manifestation de couplage entre
modes de vibrations après la capture d'un atome d'hydrogène.
- Différents mécanismes de libération d'hydrogène moléculaire. En haut nous avons
une recombinaison directe (mécanisme de Eley-Rideal). Au milieu, nous avons une
recombinaison suivant un processus "d'atome chaud". En bas nous illustrons le
dernier procédé pris en compte, la désorption induite par collision.
- Distribution du temps de vie de l'atome d'hydrogène incident sur l'agrégat avant la formation d'une molécule d'hydrogène.
- Distribution de population des niveaux vibrationnels.
- Distribution de population des niveaux rotationnels.
- Probabilités relatives des différents mécanismes de réaction avec l'hydrogène.
- Comparaison avec l'expérience.
- Evolution de la température lors d'une recombinaison et de deux absorptions
d'hydrogène atomique pour un agrégat de
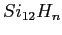 .
.
- Evolution du contenu 'n' en H d'un agrégat de
sous l'effet d'impacts
continue avec de l'hydrogène atomique.
- Distribution du contenu 'n' en H d'un agrégat de
sous l'effet d'impacts
continue avec de l'hydrogène atomique.
- Changement de structures d'un agrégat de
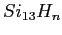 après exposition à l'hydrogène atomique.
après exposition à l'hydrogène atomique.
- Changement de structures en fonction du temps d'exposition au flux d'hydrogène atomique.
- Modification des structures après chacune des réactions successives avec un atome d'hydrogène.
A gauche les structures initiales. Nous affichons, de gauche à droite, les structures obtenues après chaque
nouvelle réaction avec un atome d'hydrogène.
A droite nous remarquons que les structures sont mieux ordonnées lorsque le nombre d'atomes de Si
est un nombre magique.
Si le nombre d'atomes de silicium de la structure est proche
d'un nombre magique, nous voyons une structure stable se former et des atomes de silicium qui ne trouvent
pas de place dans cette sous-structure stable (par exemple
).
- Illustration de quelques structures particulières obtenues lors des nos simulations.
(Fig a) structure amorphe, (Fig b) une des structures de minimum d'énergie obtenue avec un faible
flux d'hydrogène atomique identique à celle prédite par Andreoni et al. [1],
(Fig c) structure typique obtenue lors de reactions avec un haut flux d'hydrogène
atomique.
(Fig d) structure tubulaire de plus bas minimum d'énergie obtenue avec un flux intermédiaire
d'hydrogène atomique.
- Nous vérifions la stabilité d'un agrégat
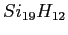 en regardant les modifications de
structure sous l'effet de la température.
La structure, qui est toujours stable à 1200 K, est détruite très rapidement à 2000 K;
(a) chauffé à 1200 K pendant 1000 ps,
(b) chauffé à 2000 K pendant 12 ps et
(c) chauffé à 2000 K pendant 15 ps.
en regardant les modifications de
structure sous l'effet de la température.
La structure, qui est toujours stable à 1200 K, est détruite très rapidement à 2000 K;
(a) chauffé à 1200 K pendant 1000 ps,
(b) chauffé à 2000 K pendant 12 ps et
(c) chauffé à 2000 K pendant 15 ps.
- (a) Densité électronique calculée avec une méthode "Self-Consistent Field".
L'échelle, donnée en unités atomique, correspond à un moment dipolaire
de 1.9 Debye.
(b) Contour selon une coupe longitudinale.
(c), (d), et (e) Contours selon des coupes transversales.
- Un empilement de deux structures tubulaires de minimum d'énergie forme une structure qui est elle
aussi une structure de minimum d'énergie.
- Vue globale du programme de lancement de réactions chimiques successives