suivant: Proportions des mécanismes monter: Mécanismes de réactions avec précédent: Capture de l'atome d'hydrogène Table des matières
La libération d'hydrogène moléculaire de l'agrégat peut avoir lieu sous plusieurs
formes. Nous avons choisi de traiter ici tous ces procédés en même temps.
Le processus le plus simple est la recombinaison directe, ou processus
de Eley-Rideal. Dans ce cas la recombinaison prend effet en quelques centaines de
femtosecondes c'est à dire le temps d'une ou deux oscillations de la
liaison Si-H temporairement formée.
La recombinaison peut aussi prendre nettement plus de temps.
En effet la barrière d'activation d'une réaction de transfert de la liaison d'un atome
d'hydrogène entre un atome de silicium vers un autre atome de silicium devient très probable
à partir d'une température de 1000 K.
Ainsi, un atome d'hydrogène peut passer facilement d'un atome de silicium à l'autre,
à condition qu'une liaison pendante se déplace dans le sens contraire.
Nous pouvons constater qu'il existe plusieurs procédés qui peuvent
mener à la formation d'une molécule d'hydrogène (Fig 5.2).
L'atome d'hydrogène incident peut réagir par un processus direct pour
former une molécule d'hydrogène [83], ou il peut au contraire commencer par un
processus d'adsorption, puis bouger sur la surface en sautant de
liaison pendante en liaison pendante, jusqu'à passer sur un atome de silicium
portant un autre atome d'hydrogène et former alors une molécule
d'hydrogène dans un processus dit "d'atome chaud" [84].

|
A partir de nos trajectoires, nous pouvons déduire le temps de vie de l'atome d'hydrogène
sur la surface de l'agrégat de silicium, et la présence ou
absence de cet atome d'hydrogène incident dans la molécule formée.
De cette façon, nous avons la possibilité
de connaître les proportions des différents mécanismes de recombinaisons induits
par l'impact d'atomes d'hydrogène.
|
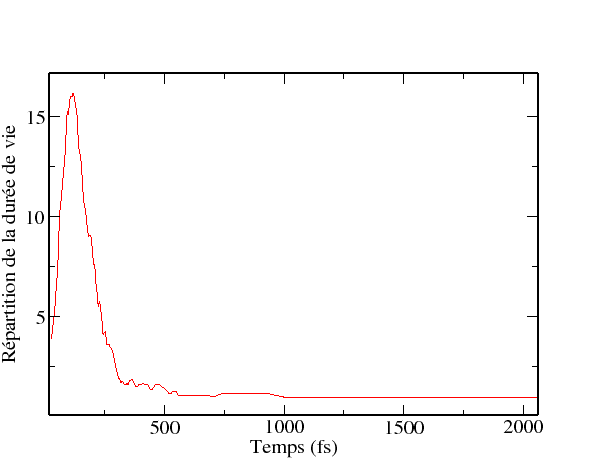
Nous avons donc analysé le temps de présence de l'atome d'hydrogène incident sur la surface de l'agrégat dans nos simulations(Fig 5.3). Pour tracer la courbe, nous avons supposé que les agrégats étaient approximativement sphériques, et nous avons créé un fichier qui contenait les temps de passages (entrée et sortie) par la sphère de diamètre égale à la plus grande distance entre des atomes de l'agrégat. Cette sphère est donc considérée ici comme la limite de l'agrégat.
Nous remarquons sur notre courbe des différences de temps de présence de l'atome d'hydrogène incident que le procédé de recombinaison est un processus très rapide dans le cas de nos agrégats. Nous pouvons faire la comparaison entre le temps que prend une vibration atomique typique de la liaison Si-H, et le temps de présence de l'atome d'hydrogène incident sur l'agrégat. En effet, sachant que la vibration d'une liaison prend environs 100 fs , nous constatons que le processus est essentiellement un processus de Eley-Rideal, c'est à dire un processus direct. Nous considérons qu'un atome d'hydrogène ayant un temps de résidence inférieur à 400 fs obéit au mécanisme d'Eley-Rideal. Quand ce temps est supérieur à 400 fs, il obéit au mécanisme dit de "l'atome chaud" ( cf Fig 5.3). Suvant ces considérations, nous trouvons un rapport de 15.9 % de molécules suivant un mécanisme d'atome chaud, pour 84.1 % de molécules d'hydrogène pour un processus de Eley-Rideal.
Nous remarquons aussi sur la distribution (Fig 5.3) que des molécules d'hydrogène quittent encore la surface de l'agrégat après 1ps. La distribution du temps de vie de l'atome d'hydrogène incident sur l'agrégat montre donc que la probabilité de désorber la molécule d'hydrogène dans un processus d'atome chaud ne se fait pas avec un temps de résidence prédéterminé. La figure 5.3 montre une probabilité de désorption constante à partir de 600 fs, mais il est impossible d'affirmer si la simulation montrerait une probabilité de désorption non nulle au bout d'un temps très long. Dans ce cas, nous pourrions supposer que l'atome d'hydrogène incident, une fois fixé sur la surface de l'agrégat, peut subir une recombinaison de type "atome chaud" après un nombre indéfini de sauts sur des atomes de silicium.
Nous considérerons par la suite les deux mécanismes, à savoir le mécanisme d'Eley-Rideal et le mécanisme d'atome chaud, comme un seul et même mécanisme de "recombinaison directe" car il met toujours en jeu l'atome d'hydrogène incident avec l'un des atomes d'hydrogène de l'agrégat. La molécule d'hydrogène issue de cette recombinaison peut être étudiée isolément.
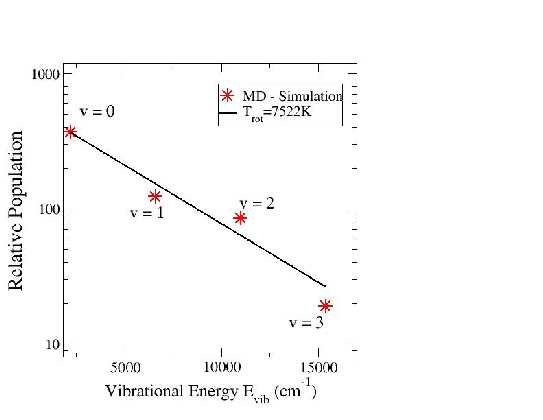
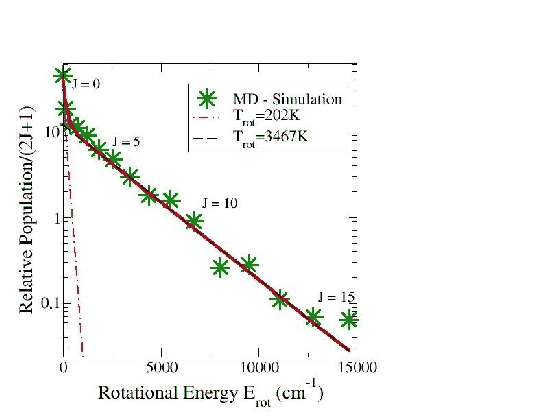
Lorsque l'atome incident réagit pour former une molécule d'hydrogène avec un autre atome d'hydrogène de l'agrégat, nous avons cherché à voir quels étaient les excitations rotationnelles et vibrationnelles de cette molécule. Nous avons repris les simulations des trajectoires de toutes les molécules d'hydrogène qui provenaient de recombinaisons entre l'atome d'hydrogène incident et l'un des atomes d'hydrogène de l'agrégat. Nous avons ensuite conservé les positions et quantités de mouvement des molécules d'hydrogène formées pour lancer des simulations indépendantes. Ces simulations nous ont permis d'obtenir les distributions de populations de molécules sur les différents nivaux d'excitation vibrationnels (Fig 5.4) et rotationnels (Fig 5.5) sur un ensemble de 600 molécules d'hydrogène.
Sur ces courbes nous voyons, en observant ces deux distributions, que les molécules d'hydrogène se placent pour la plupart dans l'état de vibration V=0 et dans l'état de rotation J=4. On remarque que notre mesure de la température nous montre un processus fortement non-boltzmannien. De plus, la température de vibration des molécules désorbées de l'agrégat est très haute (7522 K). Pour la population des niveaux rotationnels, on remarque l'apparition de deux températures différentes (202 K et 3467K). Ces propriétés ont déjà été observées lors d'expériences de réactions entre de l'hydrogène atomique et des surfaces de différentes orientations [85,86,87].