suivant: Nombres magiques des agrégats monter: Changement de structure des précédent: Changement de structure des Table des matières
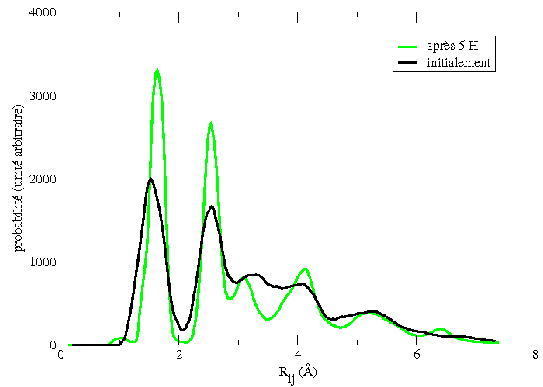
Le tracé de la fonction de corrélation de paires, pour un agrégat avant et après l'influence de l'hydrogène atomique, montre que les agrégats se cristallisent. Nous pouvons aussi remarquer que les deux maxima principaux au distances des liaisons Si-H et Si-Si. Nous voyons aussi que la forme de cette fonction montre des maximums secondaires mieux définis après une exposition à l'hydrogène atomique. Ceci montre donc que les agrégats se cristallisent après cette exposition (Fig 5.11).
REMARQUE : La fonction de corrélation de paires est le tracé de toutes les distances inter-atomiques moyennées sur un certain intervalle de temps. Nous pouvons donc déduire que la largeur des maxima est un autre indicateur de la température de l'agrégat. En effet plus l'agrégat est chaud, plus les atomes vibrent et donc plus la distance entre les voisins varie fortement. Les maximums sont donc plus larges.
Nous avons ensuite décidé de tracer la fonction de corrélation de paires dans
une fenêtre glissante dans le temps
pour voir les changements de structure qui apparaissent après chaque impact
avec des atomes d'hydrogène.
C'est à dire que nous plaçons une fenêtre de 20 fs dans laquelle nous calculons la
moyenne de la fonction de corrélation de paires de la structure. Nous déplaçons
ensuite la fenêtre de 20 fs pour calculer une
nouvelle moyenne de la fonction de corrélation de paires, ceci nous permet
d'obtenir une nouvelle courbe. En recommençant de
fenêtre en fenêtre, nous obtenons un ensemble de courbes de
fonction de corrélation de paires ordonnées dans le
temps. Ceci permet de voir, en fonction du temps, la cristallisation de l'agrégat.
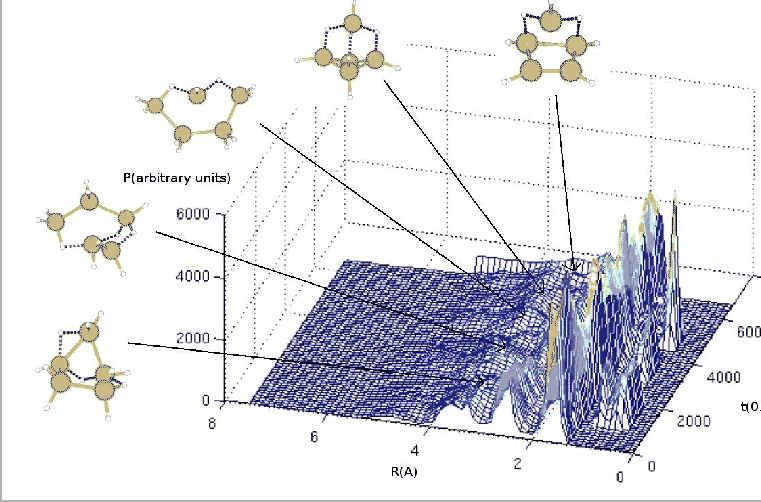
Ainsi, nous pouvons en incrémentant le temps pour chacune des courbes, obtenir un
graphique tridimensionnel qui représente la fonction de corrélation de paires en fonction du temps.
La courbe obtenue montre que la structure ne se cristallise pas systématiquement,
mais que certains impacts rendent la structure mieux ordonnée et d'autres l'amorphisent.
Donc, en visualisant les structures au cours du temps, nous remarquons que celles-ci se
modifient impact après impact. Dans le cas d'un agrégat de ![]() par exemple,
nous voyons apparaître alternativement une structure pyramidale et une structure en
anneau (Fig 5.12).
par exemple,
nous voyons apparaître alternativement une structure pyramidale et une structure en
anneau (Fig 5.12).
Nous pouvons donc dire que les structures alternativement formées sont des isomères très proches en énergie potentielle [101]. Le passage d'une structure à une autre peut donc se faire avec les variations d'énergie potentielle de la structure que la présence d'un atome d'hydrogène peut apporter lors de la formation et/ou de la destruction de liaisons. Nous pouvons comprendre ce résultat en pensant au nombre de coordination de l'atome de silicium récepteur de l'atome d'hydrogène incident. En effet, comme nous l'avons dit précédemment, selon le nombre de coordination de l'atome de silicium, des liaisons se modifient entre les autres atomes de silicium de la structure. Ainsi, la structure peut être plus ordonnée si l'atome de silicium récepteur augmente la force de ses liaisons avec les autres atomes de la structure. En effet, plus les atomes de silicium de la structure sont liés entre eux, plus l'agrégat est proche de la structure de minimum d'énergie qui est cristallisée. Les structures de minimum d'énergie potentielle sont donc les structures les plus compactes [102]. Ceci n'empêche pourtant pas d'avoir des transitions entre structures sphérique et allongée [103].
|