suivant: Modèle de croissance d'une monter: L'équation de Hartree-Fock Roothaan précédent: Méthodes paramétriques Table des matières
Nous avons maintenant trouvé une façon de calculer rapidement l'hamiltonien,
cependant nous ne savons toujours pas quelle est la fonction d'onde qui
correspond au minimum d'énergie. Pour la trouver, nous utilisons une méthode itérative
dite "méthode du champ auto-cohérent". Celle-ci consiste à se donner
empiriquement une certaine projection de la fonction d'onde atomique
sur la base LCAO décrite précédemment. Cette fonction d'onde permet une évaluation
des éléments de la matrice de Fock ![]() et donc permet de déterminer un
ensemble initial de valeurs de
et donc permet de déterminer un
ensemble initial de valeurs de ![]() pour calculer le déterminant séculaire
pour calculer le déterminant séculaire
![]() . Ainsi, on trouve une valeur de l'énergie
. Ainsi, on trouve une valeur de l'énergie ![]() de l'orbitale
moléculaire
de l'orbitale
moléculaire ![]() et donc une nouvelle décomposition de la fonction d'onde
donnée par un ensemble de coefficients
et donc une nouvelle décomposition de la fonction d'onde
donnée par un ensemble de coefficients ![]() . Ces nouveaux coefficients permettent
de se donner une ré-évaluation des éléments de la matrice de Fock et de
répéter la procédure jusqu'à atteindre une variation sur l'énergie
suffisamment faible. Une fois que cette variation est
faible, nous pouvons admettre que les coefficients de décomposition
représentent bien la projection de la fonction d'onde correspondante à un minimum
d'énergie. Pour nos simulations nous avons pris un critère d'arrêt de
. Ces nouveaux coefficients permettent
de se donner une ré-évaluation des éléments de la matrice de Fock et de
répéter la procédure jusqu'à atteindre une variation sur l'énergie
suffisamment faible. Une fois que cette variation est
faible, nous pouvons admettre que les coefficients de décomposition
représentent bien la projection de la fonction d'onde correspondante à un minimum
d'énergie. Pour nos simulations nous avons pris un critère d'arrêt de
![]() .
De cette façon, nous arrêtons la minimisation de l'énergie lorsque la variation sur
l'énergie, sur une itération, est inférieure à notre critère d'arrêt.
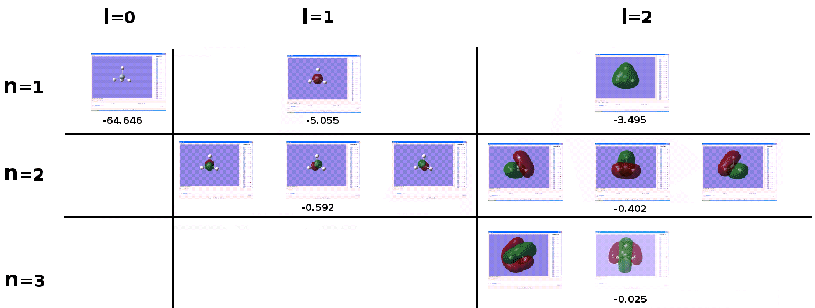
Nous pouvons ainsi voir la distribution spatiale des fonctions d'onde associées
à chaque niveau énergétique. Nous remarquerons en visualisant les différentes fonctions
d'onde électroniques, que celles des molécules peuvent être des
fonctions compliquées de l'espace (Fig 2.5).
En connaissant la répartition spatiale des électrons nous pouvons
déduire la force qu'ils vont engendrer sur les noyaux et nous pouvons utiliser
les algorithmes classiques
de trajectoires que nous avons décrit au début de ce chapitre.
.
De cette façon, nous arrêtons la minimisation de l'énergie lorsque la variation sur
l'énergie, sur une itération, est inférieure à notre critère d'arrêt.
Nous pouvons ainsi voir la distribution spatiale des fonctions d'onde associées
à chaque niveau énergétique. Nous remarquerons en visualisant les différentes fonctions
d'onde électroniques, que celles des molécules peuvent être des
fonctions compliquées de l'espace (Fig 2.5).
En connaissant la répartition spatiale des électrons nous pouvons
déduire la force qu'ils vont engendrer sur les noyaux et nous pouvons utiliser
les algorithmes classiques
de trajectoires que nous avons décrit au début de ce chapitre.
|