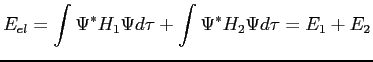suivant: Le déterminant séculaire monter: Simulation de la dynamique précédent: Combinaison linéaire d'orbitales atomiques Table des matières
Nous pouvons décomposer l'hamiltonien électronique comme la somme de deux termes. L'un pour décrire l'effet que ressentent les électrons sous l'influence des noyaux (élaboré par Hartree) et l'autre terme pour l'effet que ressentent les électrons sous l'influence des autres électrons (élaboré par Fock):
| (2.39) | |||
 |
(2.40) | ||
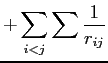 |
(2.41) |
Dans la suite nous poserons
![]() et nous omettrons les bornes
sur les intégrales qui porterons toujours sur tout l'espace.
Nous obtenons ainsi
et nous omettrons les bornes
sur les intégrales qui porterons toujours sur tout l'espace.
Nous obtenons ainsi
qui devient, en remplaçant ![]() par son expression sous la forme d'un
déterminant de Slater, et compte tenu de l'orthogonalité et du fait que
les fonctions d'onde sont normées :
par son expression sous la forme d'un
déterminant de Slater, et compte tenu de l'orthogonalité et du fait que
les fonctions d'onde sont normées :
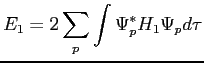 |
(2.43) |
![$\displaystyle E_2=2\sum_p\sum_q[\int\Psi^*_p(i)\Psi^*_q(j)H_2\Psi_p(i)\Psi_q(j)...
..._j -\frac{1}{2}\int\Psi^*_p(i)\Psi^*_q(j)H_2\Psi_p(j)\Psi_q(i) d\tau_i d\tau_j]$](img152.png) |
(2.44) |
L'énergie électronique totale devient donc la somme de ces deux énergies. En introduisant la décomposition des fonctions d'onde électroniques sur une base de fonctions d'onde atomiques (LCAO) :
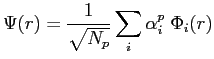 |
(2.45) |
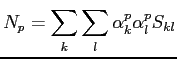 |
(2.46) |
nous obtenons
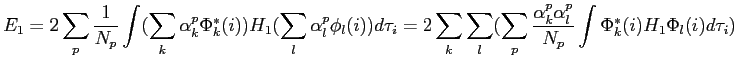 |
(2.47) |
Nous noterons par la suite
![]() et donc nous pouvons
réécrire
et donc nous pouvons
réécrire ![]() comme :
comme :
 |
(2.48) |
De la même façon :
 |
(2.49) | ||
![$\displaystyle \frac{1}{2} \int \Phi^*_k(i)\Phi^*_m(j) H_2 \Phi_l(j) \Phi_n(i) d\tau_i d\tau_j]$](img168.png) |
que nous renoterons aussi
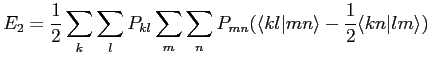 |
(2.50) |
Cette équation est la partie maîtresse de notre modélisation. Elle est connue sous le nom d'équation de Fock-Roothaan Hall [43,44] et va nous servir à déterminer la décomposition de nos fonctions d'onde électroniques sur notre base d'orbitales atomiques et donc la densité de probabilité de présence des électrons autour des atomes. L'utilisation d'une base de fonctions d'onde de type Slater, c'est à dire de fonctions d'onde obtenues par calcul analytique des orbitales de l'atome d'hydrogène, est une très bonne approximation dans le cas où les mouvements des électrons ne sont pas corrélés avec les mouvements des noyaux.