Dans notre modèle 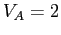 et
et  , nous fixons à partir d'une position donnée
une série d'atomes identiques A (respectivement B) en forçant le critère énergétique.
Le système, laissé libre de s'organiser par lui même, replace son niveau de Fermi
dans le gap en fixant
des atomes B (respectivement A), de façon à ce
qu'il y ait exactement autant d'atomes A que d'atomes B.
Ceci nous fait
penser à regarder s'il existe une organisation par blocs. En effet, les
chaînes s'organisent pour tout
, nous fixons à partir d'une position donnée
une série d'atomes identiques A (respectivement B) en forçant le critère énergétique.
Le système, laissé libre de s'organiser par lui même, replace son niveau de Fermi
dans le gap en fixant
des atomes B (respectivement A), de façon à ce
qu'il y ait exactement autant d'atomes A que d'atomes B.
Ceci nous fait
penser à regarder s'il existe une organisation par blocs. En effet, les
chaînes s'organisent pour tout  (même
lorsque
(même
lorsque  ce qui prouve que c'est un effet purement
électronique) en blocs de deux atomes AB ou BA, ce qui revient, comme nous
l'avons vu précédemment, à positionner le niveau de Fermi juste sous le gap.
La perturbation apportée à notre système est relaxée de façon à
conserver cette neutralité. Donc après 10 atomes A, notre système se
relaxe en ajoutant 10 ou 11 atomes B (10 si le système repart
avec un bloc AB et 11 s'il repart avec un bloc BA).
Donc nous remarquons
qu'une bonne façon de caractériser notre système est, du fait de la
neutralité par blocs, de repérer les retournements de blocs.
Par retournement de blocs nous entendons un
passage de ABABAB à BABABA. Nous pourrons donc caractériser la chaîne par
les positions où nous obtenons un passage AA ou un passage BB, trahissant un retournement
de bloc.
Nous pouvons réécrire notre chaîne où nous avions posé dans notre programme, pour la position
ce qui prouve que c'est un effet purement
électronique) en blocs de deux atomes AB ou BA, ce qui revient, comme nous
l'avons vu précédemment, à positionner le niveau de Fermi juste sous le gap.
La perturbation apportée à notre système est relaxée de façon à
conserver cette neutralité. Donc après 10 atomes A, notre système se
relaxe en ajoutant 10 ou 11 atomes B (10 si le système repart
avec un bloc AB et 11 s'il repart avec un bloc BA).
Donc nous remarquons
qu'une bonne façon de caractériser notre système est, du fait de la
neutralité par blocs, de repérer les retournements de blocs.
Par retournement de blocs nous entendons un
passage de ABABAB à BABABA. Nous pourrons donc caractériser la chaîne par
les positions où nous obtenons un passage AA ou un passage BB, trahissant un retournement
de bloc.
Nous pouvons réécrire notre chaîne où nous avions posé dans notre programme, pour la position 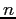 ,
la valeur
,
la valeur 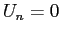 pour les
atomes B et
pour les
atomes B et 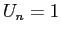 pour les atomes A (attention U représente A ou B et n'a pas d'autre signification
que le type de l'atome).
En créant une nouvelle
chaîne telle que
pour les atomes A (attention U représente A ou B et n'a pas d'autre signification
que le type de l'atome).
En créant une nouvelle
chaîne telle que
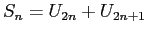 , pour mettre les atomes par deux, nous obtenons une chaîne uniquement
constituée de '1' ce qui vérifie la neutralité par blocs. De même si nous créons une
chaîne telle que
, pour mettre les atomes par deux, nous obtenons une chaîne uniquement
constituée de '1' ce qui vérifie la neutralité par blocs. De même si nous créons une
chaîne telle que
 nous obtenons de la même façon
une chaîne constituée de '1' sauf aux moments des retournements de blocs de type
nous obtenons de la même façon
une chaîne constituée de '1' sauf aux moments des retournements de blocs de type
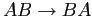 et nous obtiendrons un '2' lors d'un retournement
et nous obtiendrons un '2' lors d'un retournement
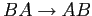 .
.
De cette façon nous remarquons que les retournements par blocs sont de moins en moins
fréquents au fur et à mesure que nous nous éloignons de la perturbation.
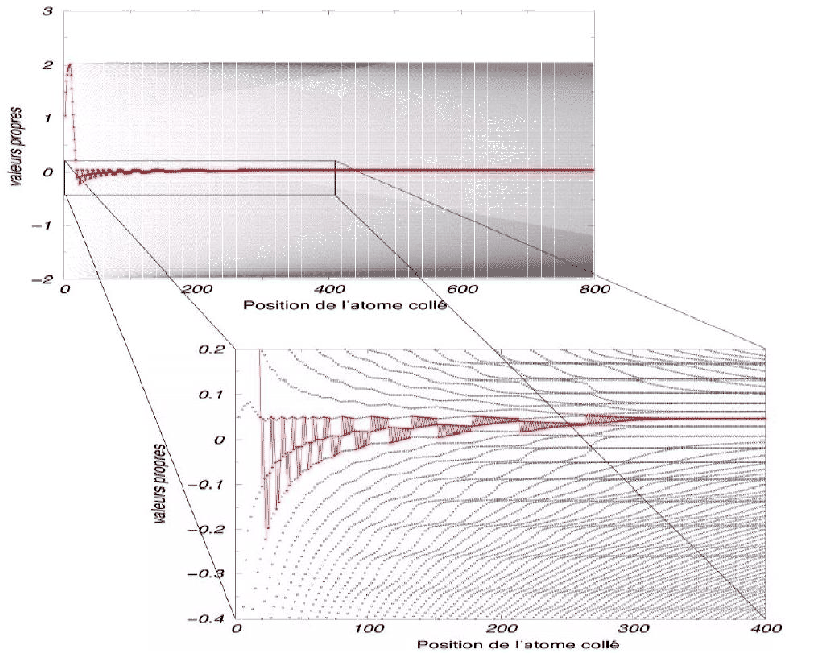
Nous pouvons le voir sur un diagramme identique à celui de la figure 3.3.
En commençant par fixer
arbitrairement 10 atomes A, nous créons des niveaux occupés et le niveau de Fermi
monte pour chaque nouveau niveau dans le diagramme (Fig 3.4).
Nous pouvons le voir sur la figure,
le niveau de fermi se replace spontanément sur les niveaux du centre par
une auto-organisation de la chaîne en AAAAAAAAAABBBBBBBBBB. Nous pouvons donc
voir le niveau de Fermi (en rouge) redescendre jusqu'au vingtième atome collé.
Nous voyons ensuite le système s'organiser en une alternance de séquences
ABABABABA et BABABABAB de plus en plus longues. La perturbation initiale
n'a plus d'effet sur les atomes collés suffisamment loin. Le programme ne crée plus de
retournement de blocs après une certaine longueur de chaîne.
Nous remarquons aussi que la perturbation perd son effet d'autant plus vite
que l'intégrale de saut '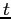 ' est petite en traçant les mêmes diagrammes pour
des valeurs différentes.
' est petite en traçant les mêmes diagrammes pour
des valeurs différentes.
Figure 3.4:
Les retournements de blocs sont de moins en moins fréquents lorsque nous nous
éloignons de la perturbation initiale (germe de croissance AAAAAAAAAA)
|
|
Ainsi, nous remarquons que pour une longueur de chaîne de plus en plus élevée,
nous perdons l'information d'un retournement de blocs avec la distance à celui-ci.
Nous remarquons en effet qu'un système initialement contraint perd l'effet de
cette contrainte au fur et à mesure de la croissance [52,53].
Nous pouvons donc déduire que,
dans le plasma, la croissance de nos agrégats ne sera pas influencée par le germe
initial. Nous verrons en effet cet effet par la suite lors de notre étude
de la dynamique moléculaire du chapitre 4.