suivant: Comparaison avec les résultats monter: Croissance des nanocristaux de précédent: Croissance des nanocristaux de Table des matières
Nous avons passé en revue tous les outils nécessaires pour
faire le calcul quantique de la structure électronique
des molécules dans le chapitre 2. Ici, nous présentons une étude dynamique, et non
plus statique, afin d'explorer la cinétique des réactions chimiques lors de
la croissance des nanostructures dans le plasma.
La plupart des études effectuées dans le passé sont surtout basées sur les
structures de minimum d'énergie [54,55,56],
cependant ces structures ne représentent pas toujours la réalité.
Pour effectuer l'étude dynamique, nous pourrions déduire le mouvement des atomes
par des méthodes totalement quantiques, c'est-à-dire en résolvant l'équation
de Schrödinger dépendante du temps
![]() mais nous serions limités par des temps de calcul qui nous confineraient
à des systèmes de quelques atomes.
Pour étudier de plus grands systèmes, nous avons opté pour une
méthode classique de calcul de trajectoires pour les noyaux, couplée
à une méthode de calcul quantique de structure électronique.
Les noyaux se déplacent donc "classiquement" dans le potentiel
des électrons qui, lui, est calculé par les méthodes quantiques semi-empiriques
(PM3).
De cette façon nous n'avons pas besoin de connaître la forme du potentiel ressenti
par les atomes voisins. La force de liaison est dépendante
de l'état électronique des atomes mis en jeu.
En effet, l'enthalpie de formation pour une liaison
mais nous serions limités par des temps de calcul qui nous confineraient
à des systèmes de quelques atomes.
Pour étudier de plus grands systèmes, nous avons opté pour une
méthode classique de calcul de trajectoires pour les noyaux, couplée
à une méthode de calcul quantique de structure électronique.
Les noyaux se déplacent donc "classiquement" dans le potentiel
des électrons qui, lui, est calculé par les méthodes quantiques semi-empiriques
(PM3).
De cette façon nous n'avons pas besoin de connaître la forme du potentiel ressenti
par les atomes voisins. La force de liaison est dépendante
de l'état électronique des atomes mis en jeu.
En effet, l'enthalpie de formation pour une liaison ![]() est
différente de celle pour une liaison
est
différente de celle pour une liaison ![]() . Dans notre simulation, nous
calculons l'énergie électronique pour les différents états possibles, et
nous considérons que les électrons se placent dans l'état de minimum d'énergie.
Le type de liaison est donc déduit automatiquement par
la méthode du champ auto-cohérent, et donc les liaisons sont formées
spontanément à partir de l'état des atomes qui y participent [57].
. Dans notre simulation, nous
calculons l'énergie électronique pour les différents états possibles, et
nous considérons que les électrons se placent dans l'état de minimum d'énergie.
Le type de liaison est donc déduit automatiquement par
la méthode du champ auto-cohérent, et donc les liaisons sont formées
spontanément à partir de l'état des atomes qui y participent [57].
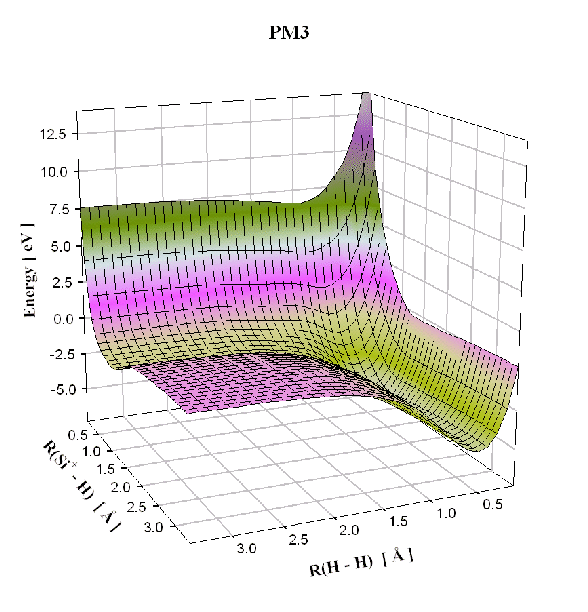
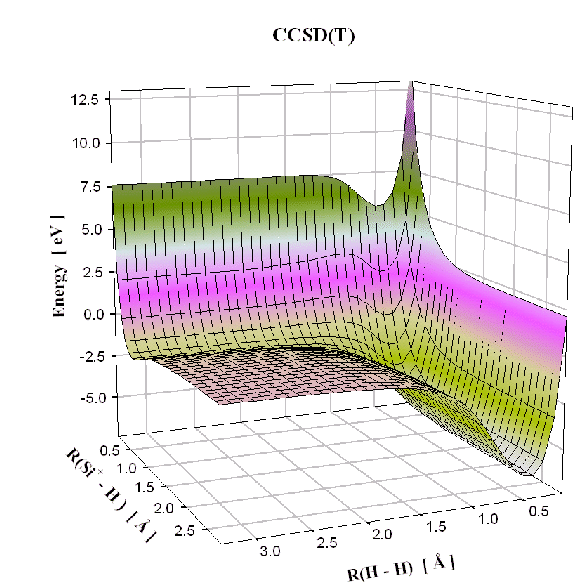
La forme des courbes de potentiels que nous utiliserons par la suite dans notre calcul de la dynamique moléculaire permet déjà de savoir si les approximations que nous avons faites précédemment sont réalistes ou totalement incohérentes dans notre cas de système silicium-hydrogène. Nous pouvons ainsi vérifier que nous avons un bon accord, étant données les approximations semi-empiriques, entre les potentiels pour des petits systèmes trouvés avec les approximations PM3, et les potentiels trouvés avec un plus haut niveau de théorie. Par plus haut niveau de théorie, nous avons pris l'exemple de la méthode "coupled clusters" (CCSD(T)) qui prend explicitement en compte les corrélations électroniques, alors que dans la simulation de type PM3 ces corrélations sont estimées à partir des données expérimentales.
|
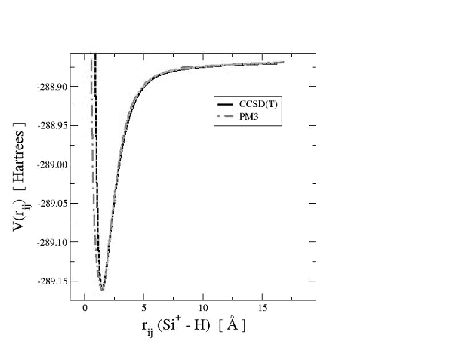
Dans la figure 4.1, nous comparons l'énergie potentielle
![]() d'une molécule Si-H obtenue avec les deux méthodes différentes:
semi-empirique PM3 et "coupled clusters". L'utilisation de la méthode "coupled clusters"
avec deux bases de fonction d'onde différentes donne des résultats similaires.
La méthode "coupled clusters" consiste à écrire la fonction d'onde comme une combinaison
de déterminants de Slater. Cette combinaison linéaire est constituée du déterminant
de Slater de la méthode de Hartree-Fock, et du déterminant de Slater des états de double
excitation des fonctions d'onde de l'état fondamental des atomes. La méthode
"coupled clusters" est donc plus précise que la méthode semi-empirique PM3.
d'une molécule Si-H obtenue avec les deux méthodes différentes:
semi-empirique PM3 et "coupled clusters". L'utilisation de la méthode "coupled clusters"
avec deux bases de fonction d'onde différentes donne des résultats similaires.
La méthode "coupled clusters" consiste à écrire la fonction d'onde comme une combinaison
de déterminants de Slater. Cette combinaison linéaire est constituée du déterminant
de Slater de la méthode de Hartree-Fock, et du déterminant de Slater des états de double
excitation des fonctions d'onde de l'état fondamental des atomes. La méthode
"coupled clusters" est donc plus précise que la méthode semi-empirique PM3.
Nous remarquons que la distance d'équilibre est presque identique. Seul le potentiel de répulsion diffère légèrement entre les deux méthodes. Cependant les réactions chimiques ont lieu à température ambiante (400K) et donc les collisions entres atomes sont suffisamment peu violentes pour que cette partie du potentiel puisse intervenir. Ainsi, le minimum de cette courbe d'énergie potentielle sera surtout important pour notre simulation, et comme nous pouvons le voir sur la figure 4.1, les minima calculés par les deux méthodes sont identiques. De la même façon nous pouvons comparer les surfaces de potentiel de la molécule