suivant: Analyse des résultats monter: Programmes développés précédent: Programme de calcul de Table des matières
Pour modéliser la croissance d'agrégats, il n'est pas possible de faire les
simulations en parallèle. En effet, il est nécessaire de connaître le résultat
d'une trajectoire pour connaître la simulation suivante. Cependant, la structure
créée pour la simulation reste bien pratique en ce qui concerne l'analyse
des résultats. Ainsi, pour chacun des ordinateurs de la grappe, nous pouvons
créer un autre succession de répertoires qui contiendront un fichier d'entrée
pour une nouvelle simulation, fabriqué en utilisant le fichier de sortie de la
simulation précédente. Ainsi, nous commençons avec un répertoire 1, dans lequel
nous simulons une réaction chimique que nous souhaitons boucler. Dans le cas
de la croissance d'un agrégat, nous simulerons une réaction entre ![]() et
et ![]() .
Nous obtiendrons un fichier de résultat qui déterminera la nouvelle réaction à effectuer
avec
.
Nous obtiendrons un fichier de résultat qui déterminera la nouvelle réaction à effectuer
avec ![]() . Par exemple, si le résultat de la réaction entre
. Par exemple, si le résultat de la réaction entre ![]() et
et ![]() est
est
![]() , nous conservons les coordonnées dans l'espace des phases de
, nous conservons les coordonnées dans l'espace des phases de ![]() pour
fabriquer un nouveau fichier d'entrée afin de simuler la réaction entre
pour
fabriquer un nouveau fichier d'entrée afin de simuler la réaction entre ![]() et
et
![]() dans le répertoire 2. De la même façon, nous allons obtenir un nouveau produit
formé lors de la réaction simulée dans le répertoire 2, qui va être utilisé pour créer un
nouveau fichier de simulation pour la troisième réaction dans le répertoire 3 et ainsi
de suite.
dans le répertoire 2. De la même façon, nous allons obtenir un nouveau produit
formé lors de la réaction simulée dans le répertoire 2, qui va être utilisé pour créer un
nouveau fichier de simulation pour la troisième réaction dans le répertoire 3 et ainsi
de suite.
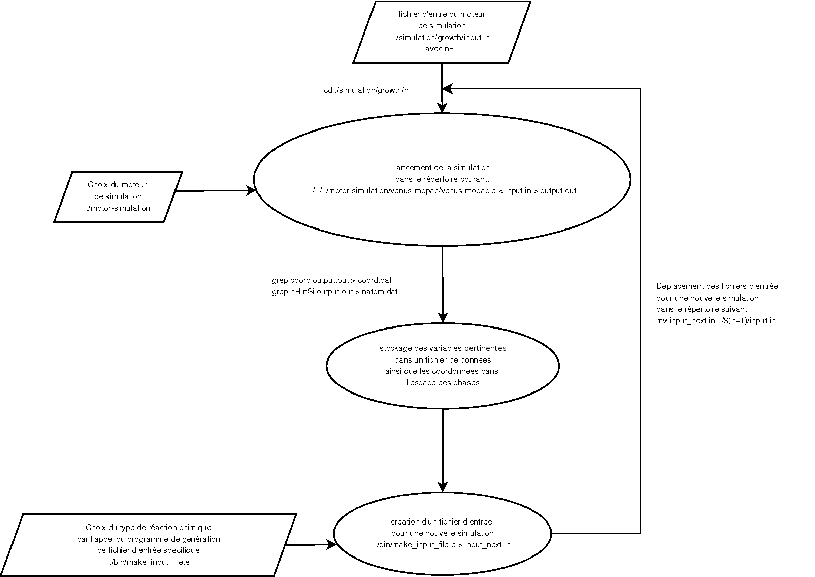
Pour créer les fichiers d'entrée pour les simulations successives, nous utilisons
les coordonnées des positions et des impulsions des molécules présentes dans le voisinage
des atomes de silicium par l'analyse de la simulation précédente. En effet, même si la
réaction de capture ne n'initialise pas dès la première réaction, elle s'initialisera
tôt ou tard. Or la propriété qu'ont les agrégats de croître autour d'un noyau de
silicium est très utile pour nous. Donc nous supposerons que le résultat d'une
simulation est un ensemble d'atomes de silicium et d'hydrogène, dans une position
voisine les uns des autres dans l'espace des phases, ainsi qu'un fragment
qui n'a pas réagi. Il faut donc déterminer quels sont les atomes qui appartiennent
à ce fragment si nous souhaitons les éliminer pour la simulation suivante.
Pour ce faire, nous calculons la position du centre de masse des atomes de silicium.
Ce centre de masse doit à priori être proche du centre de masse total de l'agrégat.
Nous pouvons trouver les molécules qui sont incluses dans une sphère de rayon arbitraire
autour de ce centre de masse, et ainsi trouver tous les atomes participant à
la formation de l'agrégat dans la simulation suivante. Nous recalculons ainsi les
coordonnées du centre de masse réelle de l'agrégat dans l'espace des phases.
Nous fabriquons ainsi un fichier d'entrée pour la simulation suivante en soustrayant
atome après atome, les positions et impulsions à la position et à l'impulsion du
centre de masse de l'agrégat formé pour immobiliser l'agrégat
au centre de l'environnement de simulation. Nous tirons ensuite aléatoirement
l'impulsion de la nouvelle molécule qui doit réagir avec l'agrégat suivant une
loi de Maxwell-Boltzmann (ou dans tout autre distribution d'ailleurs) pour placer
à nouveau les coordonnées du réactif dans le fichier d'entrée de la simulation
suivante. Nous copions ensuite ce nouveau fichier dans le répertoire suivant et nous recommençons
la procédure. Nous avons donc fabriqué un programme en C++ permettant, par des
calculs géométriques, de trouver les molécules formant l'agrégat à partir d'un
fichier de sortie de simulation, pour fabriquer un fichier d'entrée de simulation.
Pour créer ce fichier
d'entrée, nous ajoutons le réactif que nous souhaitons simuler. Ainsi, nous avons écrit un ensemble
de programmes de structures identiques qui permettent de fabriquer un fichier
d'entrée de simulation de réaction avec ![]() ,
, ![]() ,
, ![]() ,
, ![]() avec conservation
de l'impulsion obtenue lors de la simulation précédente ou en supprimant celle-ci.
Cet ensemble de 8 programmes en C++ est donc le coeur de la simulation.
En effet, les choix successifs de programmes de formation de fichier
d'entrée, pour des molécules différentes et dans des proportions définies par la composition
du milieu que nous souhaitons simuler, permet de modéliser toutes les proportions
possibles de réactions, en étant fidèle à la composition réelle du plasma, ou de tout autre
milieu réactif en phase gazeuse.
En effet, nous pouvons transposer ce programme à toute la chimie en phase gazeuse pour peu
qu'il existe des moteurs de simulations qui traduisent la réalité des réactions envisagées,
ce qui est hélas rarement le cas.
avec conservation
de l'impulsion obtenue lors de la simulation précédente ou en supprimant celle-ci.
Cet ensemble de 8 programmes en C++ est donc le coeur de la simulation.
En effet, les choix successifs de programmes de formation de fichier
d'entrée, pour des molécules différentes et dans des proportions définies par la composition
du milieu que nous souhaitons simuler, permet de modéliser toutes les proportions
possibles de réactions, en étant fidèle à la composition réelle du plasma, ou de tout autre
milieu réactif en phase gazeuse.
En effet, nous pouvons transposer ce programme à toute la chimie en phase gazeuse pour peu
qu'il existe des moteurs de simulations qui traduisent la réalité des réactions envisagées,
ce qui est hélas rarement le cas.